01
Introduction to Japan's New Drug Reexamination System
Japan established a national drug monitoring system in 1967 and established a post marketing surveillance (PMS) system in 1979 through legal means. The Japanese PMS system consists of a new drug re examination system, a drug re evaluation system, and a drug adverse reaction (ADR) reporting system.

The re review system is a systematic approach to monitor and collect the effectiveness and safety of approved new drugs from the date of approval to a certain period of time (based on different types of new drugs, with different review periods), in order to reconfirm the effectiveness and safety of new drugs on the market. Japan has strict regulations on new drugs. Although new drugs are approved for market based on clinical trials, their effectiveness and safety, especially safety, are not clear for large populations in the real world. Therefore, Japanese regulations need to reconfirm their effectiveness and safety. In 1980, the re examination system was officially established and incorporated into Japan's Pharmaceutical Law. Under this system, the approval of new drugs for marketing is only a temporary sales license, and the permanent sales qualification needs to be determined based on the re examination results.
Figure 1: Development process of Japan's new drug re examination system

02
Reexamination Implementation Method
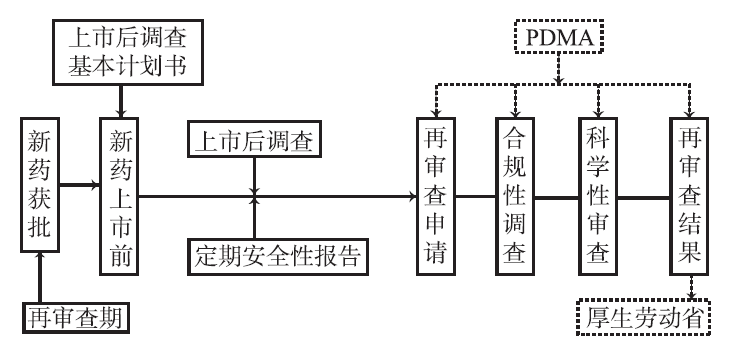
2.1 Re examination object
The re examination is supervised and implemented by the Ministry of Health, Labour and Welfare of Japan and PMDA. Generally, MAH (listing permit holder) submits a re examination application to PMDA within 3 months after the end of the re examination period and accepts the re examination. Based on different types of new drugs, the review period varies. (1) Rare disease drugs and drugs that require long-term drug epidemiological investigation for 10 years; (2) 8 years for new active ingredient drugs; (3) 8 years for new drug administration routes and new compound prescriptions; (4) 6 years for drugs with new indications, new usage/new dosage (approved drugs, only indications for orphan drugs); (5) New indications (excluding (4)) and new doses (excluding (4)) of drugs are for 4 years.
2.2 Re review content
The Pharmaceutical Affairs Law requires MAH to investigate and collect the post marketing use of new drugs and other data in the reexamination period according to the post marketing investigation standard, namely GPSP. GPSP further clarifies the post marketing investigation content, including drug use investigation and post marketing clinical trials conducted by collecting, verifying and confirming information related to drug quality, safety and effectiveness.
2.2.1 Usage survey
The main content of the usage survey is the use of new drugs in the real world, mainly referring to the medication situation in daily diagnosis and treatment within the hospital. Collect medication information from the population to evaluate effectiveness and safety. MAH collects and analyzes information related to adverse reactions, efficacy, and safety of all medication patients during the investigation of usage, in order to evaluate the safety of the drug, unknown adverse reactions, etc. You can also use a medical information database provided by a third-party medical information data service provider to investigate the post marketing medication situation. In addition, for special groups such as children and the elderly, specialized information collection surveys, called special usage surveys, are required to obtain safety information that is difficult to obtain during the research and development process.
2.2.2 Post-market clinical trials
According to regulatory requirements, if additional clinical trials (referred to as RCTs) are required, MAH needs to conduct relevant trials to evaluate the effectiveness and safety of the new drug. Similar to post market phase IV clinical trials.
Figure 2: Implementation Process of Japan's New Drug Reexamination System
03
Effect of new drug re examination
The re review system conducts routine comprehensive reviews based on the regular safety update reports and clinical trial studies of drugs after marketing. The establishment of new drug re examination in Japan can serve two purposes. On the one hand, it is to monitor the effectiveness and safety of new drugs after their launch, reconfirm their effectiveness and safety, and withdraw drugs with issues in effectiveness and safety from the market. On the other hand, re examination can grant a certain market monopoly to new drugs, and during the re examination period, PMDA will not approve the listing of generic drugs for the new drug.
04
The Enlightenment of Japan's New Drug Reexamination System on China
Japan has a strict post market monitoring system (PMS) for drugs, and post market review is an integral part of the PMS system, which focuses on monitoring new drugs after they are launched. However, China lacks a dedicated monitoring system for new drugs after their launch. The focus of new drug monitoring in China is on market monopoly, with little attention paid to the effectiveness and safety after their launch. Moreover, the new drug registration management measures no longer include a new drug monitoring system. The re examination of new drugs in Japan organically integrates post market safety data management with market exclusivity/data protection, which is worth learning from in China.
In addition, China has a pharmacovigilance system (including adverse reaction monitoring) and re registration systems for the post marketing monitoring of all drugs. However, the application of the ICH E2E guiding principle in China is still in the promotion stage, and a mature pharmacovigilance system has not yet been established. China's re registration system is relatively loose compared to Japan.
China should have a more systematic and strict monitoring system for post marketing drugs. For new drugs and marketed drugs based on different risks, supervision should be refined and treated differently to improve China's post marketing safety data management system and post marketing evaluation system. Moreover, risk management after drugs are launched cannot be solely the responsibility of drug regulatory agencies. Pharmaceutical enterprises, medical institutions, patients, and even the general public should be able to participate in the update of drug safety information in a timely manner, truly implementing the social co governance concept proposed in China's newly revised "Drug Management Law" in risk management.
Reference:
Yan Ming, Xing Hua. Drawing on the experience of CDER in the United States to improve China's drug re evaluation system [J]. International Journal of Medicine and Health, 2005,21 (1): 85-87
Ding Jinxi, Luo Xiwei, Wang Yingwei. Evaluation of Japan's Drug Data Protection System and Its Implications for China: An Empirical Study on the Performance of Japan's Innovative Drug Reexamination Policy [J]. Shanghai Pharmaceutical, 2011,32 (12): 615-620
Zhang Zhongyi, Liu Huan, Yang Yue Research on Japan's New Drug Reexamination System [J] 2019,,38(1):119-122.
Jia Guoshu, Liang Yi Research on the Postmarket Risk Management Plan of Japanese Drugs and Its Implications for China [J] China Pharmacy, 2021, 32 (19)
Lin Xiao, Wu Hongyan, et al. Research on Japan's drug vigilance system and its implications for China [J]. Chinese Journal of New Drugs and Clinical Practice, 2022,41 (5): 280-285
[Copyright Notice] Some of the images, text, and fonts in this article are sourced from the internet. The copyright belongs to the original author and is for learning and reference purposes only. Commercial use is prohibited. If there is any infringement, please contact us to delete it.
Lindmik Pharmaceutical(Suzhou)Co.,Ltd is a high-tech pharmaceutical enterprise focusing on the research and development, production and sales of innovative pharmaceutical preparations.Equipped with a number of its own innovative R&D platform of dosage forms, including the transdermal drug delivery system, and at the same time, actively introducing the world’s leading nano-based drug delivery, microspheres drug delivery and other cutting-edge pharmaceutical technologies by means of “license in”, the company is a new rapidly developing company pharmaceutical companies that catches people’s eyes.

12th Floor, Building 5, Tianyun Plaza, 111 Wusongjiang Avenue, Guoxiang Street, Wuzhong District, Suzhou City

0512-66020899

0512-66022699

215124
